"What we observe is not nature itself, but nature exposed to our method of questioning." Werner Heisenberg
AI & Representation Learning for biology
Methods that probe and quantify what protein and DNA language models actually encode.
Model- and task-agnostic evaluation of Protein/DNA embeddings' uncertainty
"All models are wrong, ..." - we formulate Random Neighbor Score (RNS) to quantify the uncertainty of
biomolecule representations like protein, DNA or RNA - in a language model’s latent space.
Publication: Prabakaran, R*., Bromberg, Y*. Quantifying uncertainty in protein representations across models and tasks. Nat Methods 23, 796–804 (2026) 13 citations
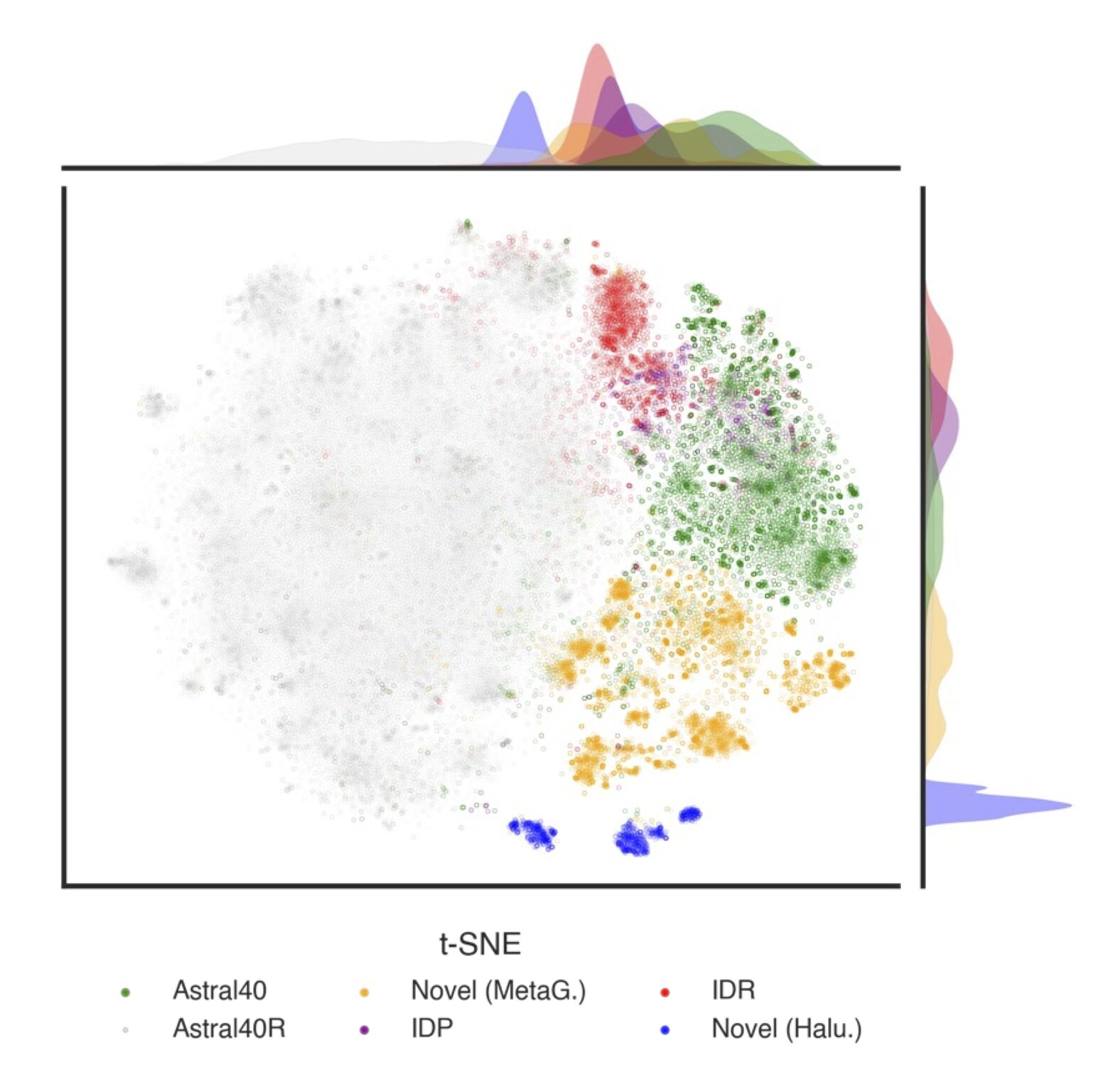
RNS with accelerated mode for protein residues and metagenomic reads Work in progress
A faster version of the Random Neighbor Score (RNS) that estimates representation uncertainty for individual protein residues and for large sets of metagenomic reads.
Linking metagenomes to the environment through representation learning Work in progress
Microbiome and environment shape each other - leading to the evolution of the biotic and abiotic world as we see today. Understanding these earth-shaping relationships is impossible without a reliable representation of the microbiome and its environment.
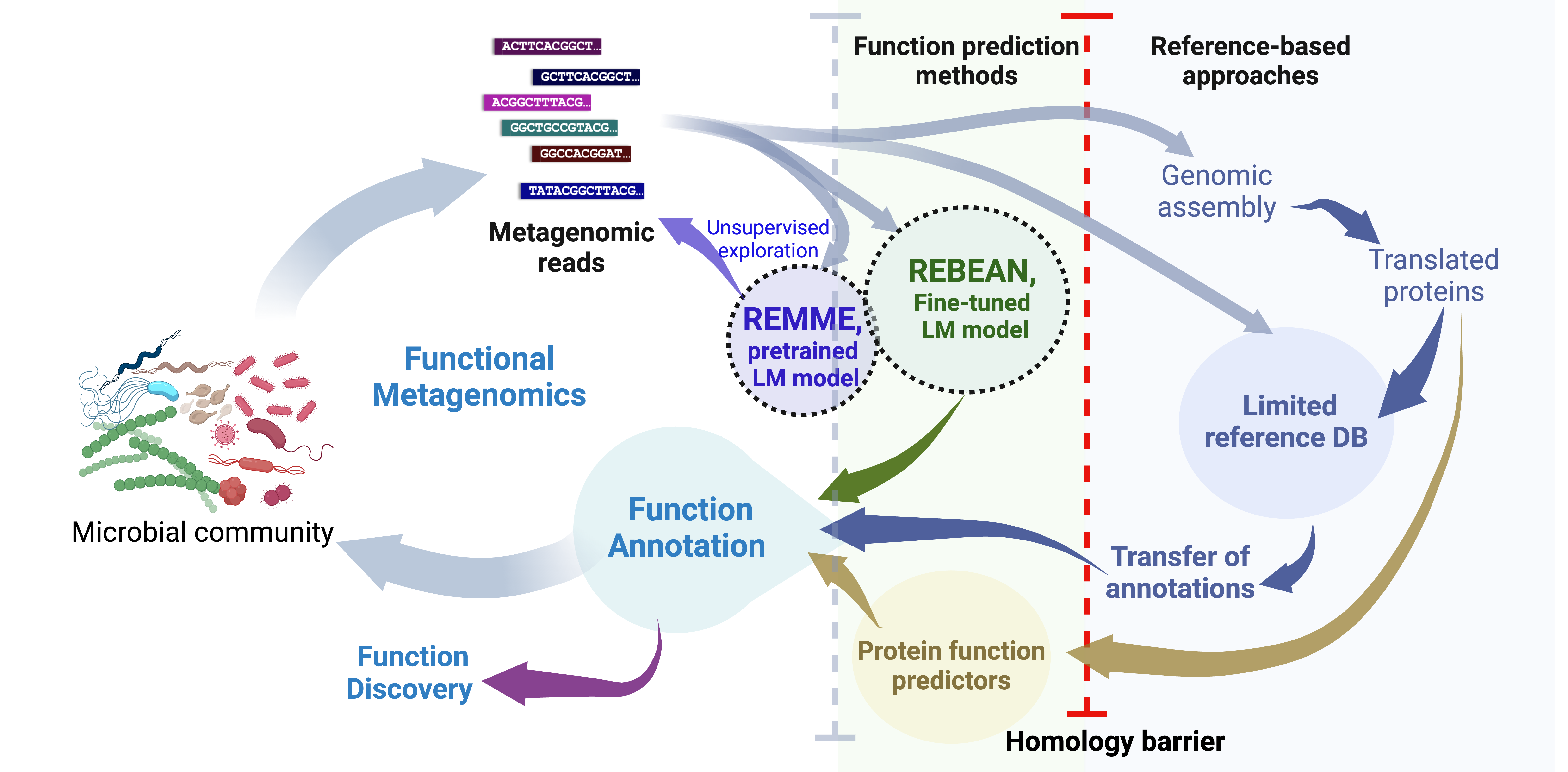
Metagenomics & reference-free function discovery
Reference-free discovery of molecular function in metagenomes using DNA language models.
Development of LMs for novel reference-free metagenomic annotation
Development of REMME, a foundational DNA language model (dLM), with a focus on biological properties of DNA, rather than only its
semantic “language-like” properties of short reads. This is crucial for analysis of
functionality of rare organisms that are significantly underrepresented in microbiomes.
To demonstrate the power and usefulness of REMME, we then developed REBEAN – a
fine-tuned model for predicting the enzymatic potential captured by individual metagenomic reads.
REBEAN does not rely on sequence comparisons, thus enabling higher coverage and faster and
more generalizable analysis of metagenomic samples, even when our understanding of the encoded
functions is limited. Using REBEAN, we mined marine metagenomes to uncover numerous novel, i.e.
previously unseen, oxidoreductases, thereby lighting a path through the microbial “dark matter.”
Publication: Prabakaran R* and Bromberg Y*, Deciphering enzymatic potential in metagenomic reads through DNA language models, Nucleic Acids Research, 53 (16), 2025, gkaf836 4 citations
NF-EnzymeFinder: end-to-end Nextflow pipeline for novel enzyme mining Unpublished
An end-to-end Nextflow pipeline that identifies enzymatic reads, assembles them into proteins, and delivers novel enzymes with predicted structures.
Source (private): https://github.com/rpkarandev/mag-rebean
Read-level AMR detection for metagenomic surveillance Work in progress
Detecting antimicrobial resistance (AMR) directly at the whole-metagenome sequencing (WMS) read level, enabling rapid metagenomic surveillance of resistance across microbial communities.
Protein function annotation & assessment
New ways to predict and rigorously evaluate protein function beyond alignment and fixed ontologies.
A protein pair-based assessment of in silico prediction tools
To address the sequencing-accelerated annotation gap, we evaluated the ability of in silico
protein function prediction tools to annotate orphan proteins, identified in metagenomes.
Lacking the “ground truth” functional annotations, we transformed the assessment
of function prediction into evaluation of functional similarity of protein pairs. This transcends
the limitations of functional annotation vocabularies, and provides a means to assess different-ontology annotation
methods.
Publication: R Prabakaran, Yana Bromberg, Functional profiling of the sequence stockpile: a protein pair-based assessment of in silico prediction tools, Bioinformatics, Volume 41, Issue 2, February 2025, btaf035 3 citations
Source: https://bitbucket.org/bromberglab/siblings-detector/
A siamese-transformer-twin model to identify protein pairs of the same function
We built a siamese model based on transformer architecture to identify protein pairs of the same function.
The model was trained using contrastive loss.
Source (private): https://bitbucket.org/bromberglab/siamtwinprot/src/main/
Variant effect prediction in the age of machine learning
A comprehensive review and assessment of variant/mutation effect predictors.
This collaborative project includes evaluation on multiple datasets
- representing different aspects of pathogenic effect.
Publication: Bromberg Y, Prabakaran R, Kabir A, Shehu A. Variant Effect Prediction in the Age of Machine Learning. Cold Spring Harb Perspect Biol. 2024 Jul 1;16(7):a041467. 30 citations
Protein sequence–structure, aggregation & evolution
Sequence- and structure-level analysis of protein aggregation and evolutionary signal.
Tracing extant metal-binding spheres in protein structural space
We searched the protein structural space for primitive, extant metal-binding spheres to trace protein evolution
Systematic study of factors contributing to peptide aggregation through extensive MD simulations
Aggregation of therapeutic monoclonal antibodies (mAbs) can negatively affect their chemistry, manufacturing,
and control attributes and lead to undesirable immune responses in patients.
The disruption of short sequence motifs called aggregation prone
regions (APRs) found in amino acid sequences of mAb candidates can potentially mitigate their aggregation.
In this work, we have performed molecular dynamics simulations to study the aggregation of an APR (VLVIY)
found in λ light chains of human antibodies and its single point mutant KLVIY. Eighteen different multicopy
peptide simulation systems of "VLVIY" and "KLVIY" were constructed by varying their concentrations, temperatures,
termini capping, and flanking gate-keeper regions.
Publication: Prabakaran, R., Rawat, P., Yasuo, N., Sekijima, M., Kumar, S., & Gromiha, M. M. (2022). Effect of charged mutation on aggregation of a pentapeptide: Insights from molecular dynamics simulations. Proteins, 90(2), 405–417. 8 citations
Development of ML and statistical models to predict protein aggregation
We studied and reviewed mechanistic models, kinetic models, ML tools and molecular dynamics approaches
used to study aggregation-prone region, aggregation propensity and aggregation rate of peptides and proteins.
This review led to development of new tools to predict aggregation prone regions and
aggregation rates: ANuPP, AggreRATE-Pred & AbsoluRATE.
Selected publications:
- Prabakaran, R., Rawat, P., Kumar, S., & Michael Gromiha, M. (2021). ANuPP: A Versatile Tool to Predict Aggregation Nucleating Regions in Peptides and Proteins. Journal of molecular biology, 433(11), 166707. 76 citations
- Prabakaran, R., Rawat, P., Thangakani, A.M. et al. Protein aggregation: in silico algorithms and applications. Biophys Rev 13, 71–89 (2021). 71 citations
Proteome-scale sequence & structural analysis of aggregation propensity
We surveyed the incidences of aggregation prone regions (APRs) in multiple proteomes
by using sequences of experimentally validated amyloid‐fibril forming peptides and via
computational prediction tools.
Publication: Prabakaran, R., Goel, D., Kumar, S., & Gromiha, M. M. (2017). Aggregation prone regions in human proteome: Insights from large-scale data analyses. Proteins, 85(6), 1099–1118. 33 citations
Other modeling work
Epidemiological and other computational modeling.
HySEIQR: Incorporation of quarantine and lockdown regulations into SEIQR model to predict COVID-19 progression
A novel hybrid SEIQR model incorporating the effect of quarantine and lockdown regulations for COVID-19
Publication: Prabakaran, R., Jemimah, S., Rawat, P. et al. A novel hybrid SEIQR model incorporating the effect of quarantine and lockdown regulations for COVID-19. Sci Rep 11, 24073 (2021). 28 citations
Webserver: https://web.iitm.ac.in/bioinfo2/covid19hyseiqr/